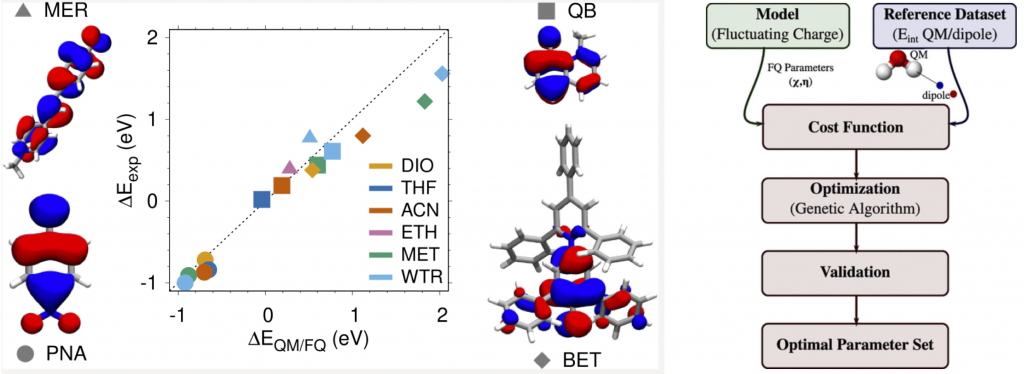
The new paper from Matteo Ambrosetti, Sulejman Skoko, Tommaso Giovannini, and Chiara Cappelli has just been published in the Journal of Chemical Theory and Computation, with the title “Quantum Mechanics/Fluctuating Charge Protocol to Compute Solvatochromic Shifts” https://pubs.acs.org/doi/10.1021/acs.jctc.1c00763.
In this work, the authors have used a genetic algorithm in the optimization of the fluctuating charges (FQ) parameters for different solvents including 1,4-dioxane, tetrahydrofuran, acetonitrile, ethanol, methanol, and water. These parameterizations were challenged to reproduce solvatochromic shifts and gave a reliable description of the experimental trends.
The extension of QM/FQ to solvents of various polarities and hydrogen-bonding capabilities opens a whole new range of possibilities of applying this technique to model the spectral properties of solvated systems.
© 2026 ERC-GEMS. Built using WordPress and the Mesmerize Theme